Available Registries for Congenital FVII Deficiency
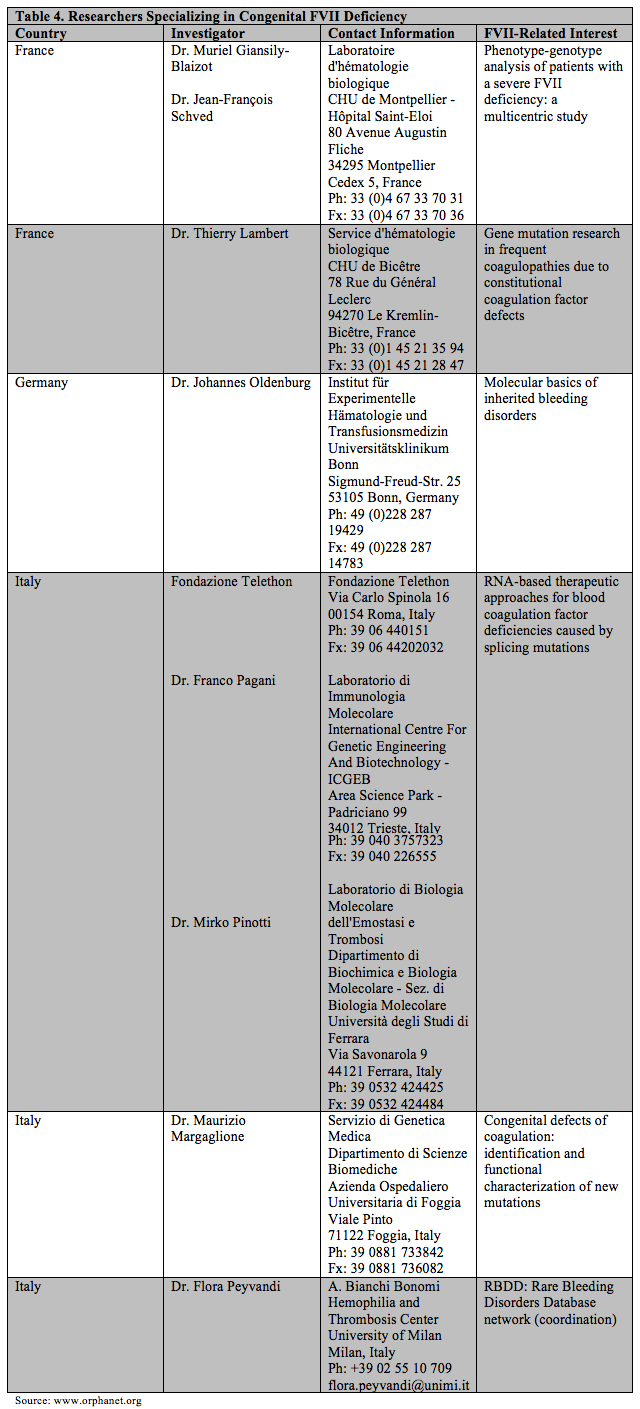
Registries, especially those that collect data prospectively, are fundamental to the efficient capture of information on orphan diseases. Such information capture is particularly applicable to the rare bleeding disorders. The retrospective International Registry on Factor VII (IRF7) was started in 1999 and closed in 2005. The Study Group for this registry achieved two main goals: (1) the establishment of a data collection system to create an efficient online registry without significant changes, and (2) an easy method of data collection that allowed for collation of information (clinical, laboratory and genetic) pertaining to 514 patients, probably representing the whole disease spectrum.
The database was used subsequently to create an electronic protocol that was the basis of the Seven Treatment Evaluation Registry (STER). This prospective registry was created in 2004 with the aim of carrying out postmarketing pharmacosurveillance for rFVIIa. The most important feature of the STER was the fact that treatment data could be analyzed with reference to the clinical and clotting phenotype of the patient, thus permitting researchers to ascertain the best treatment modality for a given clinical variant.
Table 3 summarizes the key features of the IRF7 and STER registries.
Pros and Cons of Registries
Registries are important for understanding the main features and natural history of rare disorders, and to develop treatment strategies suited to the clinical needs of the patients. Registries also provide indications for drug development and foster collaboration by creating databases of mutations to increase visibility for a rare disorder. Another benefit of registries is that they make sites or organizations available for quality control and standardization of procedures.
The disadvantages of registries are that they do not collect epidemiologic data, and even prospective data collection systems cannot compete with controlled clinical trials.
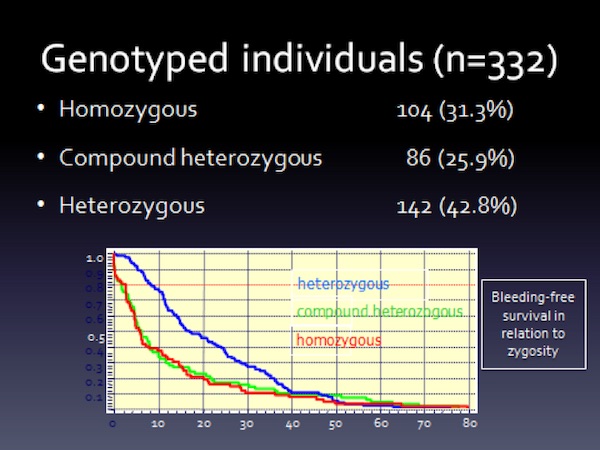
Collecting or performing genetic analysis with registry data was associated with logistic and privacy issues. The registry researchers were able to genotype 332 patients, of which 190 had changes involving both alleles. A salient aspect of the study was that the clinical spectrum of the disorder did not differ between homozygous and compound heterozygous individuals for a mutation affecting the FVII gene (Figure 7). A description of the mutational spectrum is provided in Mariani G, et al., “Clinical phenotypes and factor VII genotype in congenital factor VII deficiency”. Thromb Haemost 2005;93:481-87.
Future Research
The focus of future research in congenital FVII deficiency involves the development of an accurate bleeding score and clinically-guided treatment plans and guidelines.
Therapeutic approaches that do not involve factor replacement are being studied with the aim of improving the clinical phenotype of severely FVII-deficient patients.
Cellular models and in vivo experience with patients having severe FVII deficiency indicate that certain aminoglycosides, such as gentamicin, have the ability to increase FVIIc levels by suppressing premature termination of translation by nonsense mutations. These mutations are associated with a life-threatening bleeding tendency.45,46 Researchers continue to search for similar but more powerful molecules to obtain hemostatic levels of the missing factor.47
Researchers specializing in congenital FVII deficiency are shown in Table 4.