Factor X Deficiency
Pattern of Inheritance
Severe (homozygous) FX deficiency is inherited as an autosomal recessive disorder and is more prevalent in populations in which consanguineous marriage is common. The earliest reported molecular abnormality of the F10 gene described a female patient who was monosomic for 13q34 and was deficient in both FVII and FX.9 Her brother was trisomic for 13q34 and had increased levels of both FVII and FX.
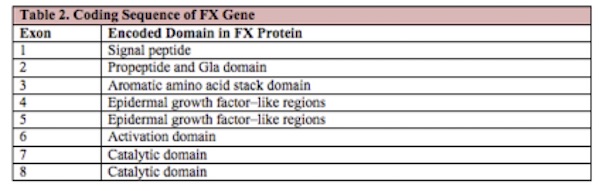
The 22 kb long FX gene (F10) is located at 13q34-ter, 2.8 kb downstream of the F7 gene. The coding sequence is homologous to that of the other vitamin K-dependent proteins and is divided into 8 exons, each of which encodes a specific domain within the protein, as shown in Table 2 below.
The F10 cDNA consists of 1474 bp coding for the pre-pro-leader sequence, the 488-amino acid mature protein, a short 3’ untranslated region, and the poly (A) tail. No TATA box has been identified in the 5’ region, allowing for multiple transcription initiation sites.
A complete absence of FX is incompatible with life, as suggested by studies with FX knockout mice with an 18-kb deletion of the F10 gene.10 The mice died of bleeding complications in utero or within the first 30 days of life, potentially explaining why severe FX deficiency is one of the rarest bleeding disorders. Livnat et al reported on 3 unrelated Palestinian patients who had severe FX deficiency with undetectable FX level (<1 U/dl). All patients were found to be homozygous for c302delG, a new frameshift mutation in the F10 gene causing a stop codon at amino acid 73.11 A new experimental model using the Zebrafish via genome editing is yielding further insights and confirming several novel human F10 mutations.12
Of the more than 130 different mutations of the F10 gene that have been identified among individuals with FX deficiency,5,13-15 most are single base pair substitutions13 and are private (i.e., unique to a particular patient or family). The Greifswald Factor X Deficiency Registry, which includes 102 patients with confirmed F10 gene mutations, identifies 29 different mutations from 45 families: 26 missense mutations, 2 microdeletions, and 1 splice site mutation.15 The most common sites of mutation have been localized to the Gla domain (exon 2) and the catalytic site (exons 7 and 8)1 (see Figure 1). While mutations are typically homozygous, combined heterozygous mutations resulting in moderate F10 deficiency have also been reported.16,17
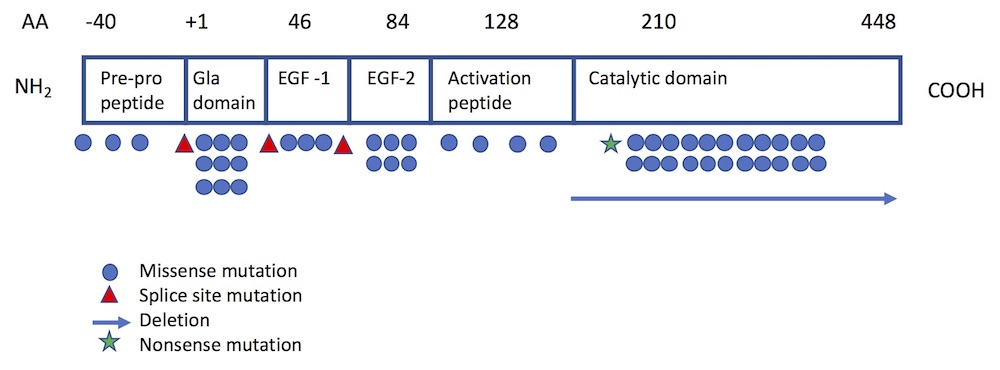
Figure 1. Location of FX mutations projected on functional protein domains.
Adapted from Perry (Haemophilia 1997) and Peyvandi et al (Haemophilia 2002).
Source: Brown DL, Kouides PA. Diagnosis and treatment of inherited factor X deficiency. Haemophilia (2008), 14, 1176–1182.
Genetic Testing Options
The website of the US National Center for Biotechnology Information (NCBI) houses the Genetic Testing Registry (https://www.ncbi.nlm.nih.gov/gtr/), which lists clinical laboratories and the clinical tests they offer (http://www.ncbi.nlm.nih.gov/gtr/conditions/C0015519/), including tests for FX deficiency.
Currently, no clinical laboratory in the United States offers genetic mutation analysis or prenatal diagnosis for FX deficiency. The Genetic Testing Registry lists laboratories across the world that conduct sequence analysis of the entire coding region (https://www.ncbi.nlm.nih.gov/gtr/). There are no available genetic databases for FX deficiency, but several useful FX mutation tables are available on the website of the International Society on Thrombosis and Haemostasis (http://c.ymcdn.com/sites/www.isth.org/resource/resmgr/publications/fx_mutations-2011.pdf ) as well as on the Human Gene Mutation Database at the Institute of Medical Genetics at Cardiff (http://www.hgmd.cf.ac.uk/ac).
Next
