
Fibrinogen is a 340 kDa complex glycoprotein synthesized in the liver that has a multitude of functions in the final steps of the coagulation cascade, including: fibrin clot formation, activated FXIII-mediated fibrin crosslinking, non-substrate thrombin binding, platelet aggregation, and fibrinolysis.1-3
The conversion of fibrinogen to insoluble fibrin is a pivotal step in the coagulation pathway. After fibrin clot formation, exposure of the non-substrate thrombin-binding sites in fibrinogen promotes antithrombotic properties.4 Therefore, as fibrinogen has multifaceted roles in coagulation, quantitative and qualitative modifications of fibrinogen can result in both bleeding and (paradoxically) venous and arterial thrombotic phenotypes.
Fibrinogen and fibrin bind and sequester thrombin, thus downregulating thrombin activity. This physiologic function was determined to be so important that fibrinogen was initially named antithrombin I. In the absence of fibrinogen and fibrin, thrombin lacks a critical regulatory pathway and is available for activation of platelets and other blood and vascular cells.5
Fibrinogen is the product of three linked genes—FGB, FGA, and FGG, ordered from centromere to telomere and are located on human chromosome 4(q31).6 These three genes encode three homologous polypeptide chains – Bb, Aa, and γ, respectively – that assemble in a sequential, pairwise manner to form mature hexameric circulating fibrinogen [(AaBbγ)2].1 The hexamer is held together by 29 disulfide bonds, and is characterized by a symmetrical structure with a central E domain that is connected to two peripheral D domains (Figure 1). Genetic mutations in the three linked fibrinogen genes give rise to a number of disorders that are known collectively as congenital fibrinogen deficiency (CFD).
Fibrin is produced by proteolytic cleavage of the fibrinogen Aa and Bβ chains by thrombin, releasing fibrinopeptides A and B (FPA and FPB, respectively) thereby allowing polymerization to occur. Cleavage of FPA (Aα 1-16) by thrombin exposes a polymerization site in the E domain that binds to the carboxyterminal region of the fibrin monomers (Figure 1).
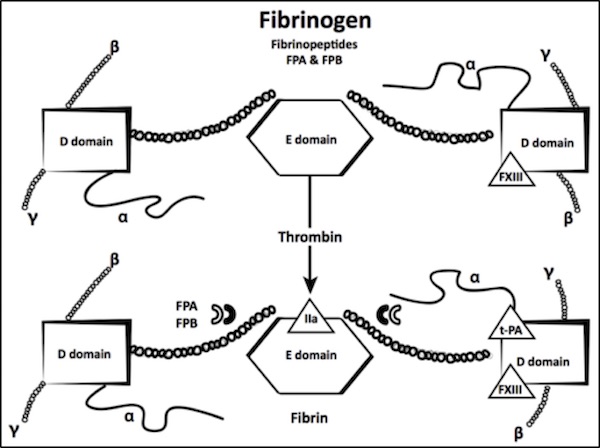
Adapted from Acharya A, Dimichele D. Rare inherited disorders of fibrinogen. Haemophilia. 2008;14:1151–1158.
FPB (Bb 1-14) cleavage occurs more slowly and contributes to lateral fibril and fiber association. Absent or slow FPB release with delayed polymerization of the fibrin monomers can cause a bleeding phenotype, while other defects in FPB release result in polymerization abnormalities that are associated with thrombotic events.
Finally, the soluble fibrin clot is stabilized by activated FXIII transglutamination to form g–g dimers and a polymers. Plasmin cleavage at sites that are located between the D and E domains in all three chains produce fragments Y, D, and E. Abnormal fibrinogens that exhibit defective crosslinking by activated FXIII have been associated with bleeding and abnormal wound healing, while abnormalities that interfere with plasminogen binding or activation on the fibrin clot result in clinical thrombosis.1
Congenital fibrinogen deficiencies (CFD) are rare inherited disorders that stem from both quantitative and qualitative defects. Quantitative defects (afibrinogenemia and hypofibrinogenemia) are characterized by reduced quantities of both the clottable protein and the immunoreactive antigen. Qualitative defects (dysfibrinogenemia) are characterized by low levels of clotting protein activity with normal or mildly-reduced antigen.
The first clinical report of bleeding due to CFD dates back to a 1920 report that described a 9-year old boy who lacked detectable fibrinogen in his blood and had suffered from recurrent bleeding episodes since birth.7 This bleeding syndrome, termed afibrinogenemia, was subsequently demonstrated to follow an autosomal recessive inheritance pattern with a variable phenotype.8,9
Based on fibrinogen assay methods, fibrinogen disorders are classified as follows:
Afibrinogenemia (homozygous or, as more commonly found in the US, compound heterozygous states) refers to the total absence of fibrinogen as measured by antigenic and functional assays. Afibrinogenemia in some reports is defined as <50 or <20 mg/dL, due to the inability of some assays to detect fibrinogen concentrations below these levels. These less-sensitive assays do not prove a total absence of fibrinogen, and probably contribute to the 25% rate of asymptomatic patients identified as afibrinogenemic.
Hypofibrinogenemia (heterozygous state, which can be confirmed only by genetic studies) is defined by a decreased level of normal fibrinogen (activity and antigen between 20 mg/dL and the lower limit of the normal range for the performing laboratory).
Dysfibrinogenemia is characterized by a structural abnormality of the fibrinogen molecule resulting in altered functional properties. Classically, the functional assay of fibrinogen yields low levels as compared with the immunological assay. However, clottable fibrinogen levels may be normal and concordant with the antigen level in some cases, such as where the abnormal fibrinogen has defective plasminogen binding with decreased fibrinolysis. The diagnosis is suspected if the ratio of fibrinogen activity to antigen is < 0.7, although this cutoff has not been prospectively validated.10
Hypodysfibrinogenemia is defined by both quantitative and qualitative defects in fibrinogen, resulting in levels ranging from 20 mg/dL to the lower limit of the normal range for the performing laboratory.
The definition and classification of CFD have changed over time with the introduction of increasingly sophisticated assay methods; hence many cases that were classified originally as hypofibrinogenemia are now classified as hypodysfibrinogenemia.
The estimated prevalence of afibrinogenemia, the most severe form of the disorder, is approximately 1 in 1,000,000.11 However, an understanding of the true incidence of these disorders has been confounded by publication bias. In populations where consanguinity is more common, as noted in the Iranian Registry, the prevalence may be similar to other autosomal recessive disorders.11,12 In fact, a 7-fold higher incidence of fibrinogen disorders was observed in the Iranian Registry for Rare Bleeding Disorders in comparison to similar registries in Italy and U.K.11
The World Federation of Hemophilia (WFH) annual global survey conducted in 2009 and the European Network of Rare Bleeding Disorders (EN-RBD) found that 7-8% of rare bleeding disorders are congenital fibrinogen disorders.13,14 The prevalence of heritable dysfibrinogenemia has been estimated as 15 per 100,000 of individuals undergoing routine coagulation laboratory testing.15
