
Plasminogen activator inhibitor-1 (PAI-1) deficiency is a rare inherited autosomal recessive bleeding disorder characterized by excessive clot lysis leading to a lifelong moderate bleeding diathesis. PAI-1 is an essential protein critical in down-regulation of the fibrinolytic pathway.
The protein: PAI-1 is a 47kDa protein and is a member of the serine protease inhibitor (SERPIN) superfamily.1,2 The PAI-1 gene in humans is located on chromosome 7q21.3-22, spans approximately 12kb, and consists of nine exons. Its expression is induced by insulin, transforming growth factor beta and endotoxin. PAI-1 is synthesized and secreted from endothelial cells, megakaryocytes, hepatocytes and adipocytes;3 it is also present in alpha granules.4 Platelets have ten times more PAI-1 antigen than present in the circulating plasma, and Simpson et al suggested that these two pools of PAI-1 are independent.5 About 80% of the PAI-1 stored in platelet alpha granules is in the latent inactive form whereas the majority of plasma PAI-1 exists in an active form.3 This may serve as an explanation for the occurrence of bleeding events in cases where only a plasma deficiency of PAI-1 is documented. The latent form can be converted to the active form with denaturants or negatively charged phospholipids in vitro. However, this conversion does not appear to occur in the circulation and clear in vivo evidence of the site of conversion is unavailable. The physiological role of the latent form has not been completely elucidated but it could participate in localized, regulatory functions of coagulation, fibrinolysis in platelet matrix interactions, and protection of the matrix against proteolysis.6 Binding of PAI-1 to vitronectin stabilizes the active form.7,8 Presently available PAI-1 antigen detection methods do not differentiate between the active and latent form.
There are reports from the 1980s regarding PAI-2 purified from placenta, leucocytes and histiocytic lymphoma cells.9-11 A study demonstrated that both PAI-1 and -2, tissue plasminogen activator (t-PA) and urokinase plasminogen activator (u-PA) increase in pregnancy despite overall fibrinolytic activity remaining constant. PAI-1 levels increased steadily after the 20th week of pregnancy and at term were three-fold higher than levels in non-pregnant women. PAI-2 levels were below the detection limit in normal plasma yet at term were 25-fold higher.12 Recent literature on PAI-1 has not differentiated between PAI-1 and PAI -2.
Physiological role of PAI-1: Fibrinolysis is a critical hemostatic process that regulates control of clot dissolution and hence wound repair, yet also plays key roles in inflammation, neoplastic and other biological processes.13,14 The major components of the fibrinolytic pathway are plasminogen activators, plasminogen, plasmin, fibrinogen, fibrin, Factor XIII, alpha-2 antiplasmin and plasminogen activator inhibitor (Figure 1).
Plasminogen activators (PA), u-PA and t-PA, are highly specific serine proteases that cleave the zymogen plasminogen, catalyzing the formation of plasmin, the primary protease essential for fibrinolysis.15 u-PA is present in plasma and other tissues while t-PA is produced by endothelial cells. Physiologic fibrinolysis occurs exclusively on the clot surface within a blood vessel and not in the circulation. The fibrin clot provides an optimal surface to increase the efficiency of plasmin generation through formation of a complex of fibrin, t-PA and plasminogen.
Regulation of fibrinolysis occurs through alpha 2-antiplasmin which is the physiologic inhibitor of plasmin, and through plasminogen activator inhibitor type-1 (PAI-1) which binds plasminogen activators in a stoichiometric manner resulting in their irreversible inhibition (Figure 1). t-PA-mediated plasminogen activation occurs prior to the dissolution of fibrin in the circulation; while u-PA binds to a specific cellular receptor (u-PAR) resulting in enhanced activation of cell-bound plasminogen. In normal plasma, t-PA activity is extremely low and the majority of t-PA is in complex with PAI-l.
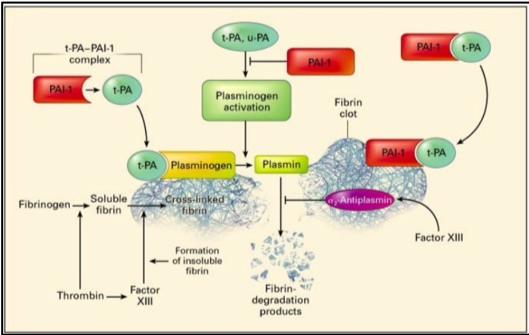
Figure 1: Plasminogen activators, urokinase plasminogen activator (u-PA) and tissue plasminogen activator (t-PA), circulate in plasma as a reversible complex with PAI-1. When the fibrin clot is formed, plasminogen and t-PA or u-PA bind to the clot and form plasmin which results in lysis of the cross-linked fibrin to fibrin degradation products. PAI-1 also binds to fibrin and when bound, can irreversibly inhibit plasminogen activators and is thereby termed a “suicide inhibitor”.16 Image from: New England Journal of Medicine, Kohler H & Grant P, Plasminogen-activator inhibitor type 1 and coronary artery disease, 2000, volume 342, p 1794. Copyright © 2000 Massachusetts Medical Society. Used with permission.
Other functions of PAI-1: Differential interactions of PAI-1 and vitronectin may influence wound healing.17,18 Plasminogen activators (PA) have been implicated in regulation of embryogenesis, angiogenesis, ovulation, inflammation, and tumor metastasis, in turn suggesting that the plasminogen activation system is an important mediator of tissue remodeling and cell migration.19 Fay et al. (1997) reported 7 homozygous children <15 years of age in an Amish kindred with complete PAI-1 deficiency,20 and hypothesized that as the identified affected population was young, PAI-1 could play an important role in human fertility; however, the clinical impact of the deficiency state in this physiologic function could not be established at that time. Animal studies performed in mice, rats, rhesus monkeys and bovine models investigating the role of the plasminogen activator system in reproduction have been reported. These data suggested that PA and PAI-1 play a crucial role in degradation of the follicular wall during ovulation. In addition to ovulation, the proteolytic process has been suggested as important in other reproductive functions including fertilization, embryo implantation and embryogenesis (please refer to “Special consideration in women” section for details).21 However, this remains controversial as a study by Carmeliet et al in PAI-1 -/- mice indicated that maternal PAI-1 may not be required for normal embryonic development.22,23 PAI-1 is also abundant in extracellular matrix and may regulate proteolysis in the extravascular space by inhibiting plasminogen activators (PA) and secondary target proteases such as thrombin.24-27 PAI-1 -/- mice are resistant to the development of pulmonary fibrosis after lung injury, also supporting the regulation of extravascular processes by PAI-1.28
PAI-1 plays an important role in aging and is secreted in senescent cells. Prolonged lifespan has been demonstrated in the PAI-1 deficient murine model.29 Berne, Indiana, is home to the largest kindred with the loss of function mutation on the PAI-1 gene, SERPIN1 (c.699_700dupTA). In a recently conducted NIH funded study, 177 Amish members of this kindred were enrolled, including 10 homozygotes, 108 heterozygotes and it was found that heterozygosity for PAI-1 was associated with longer leucocyte telomere length, lower rate of diabetes, lower fasting insulin levels and greater lifespan.
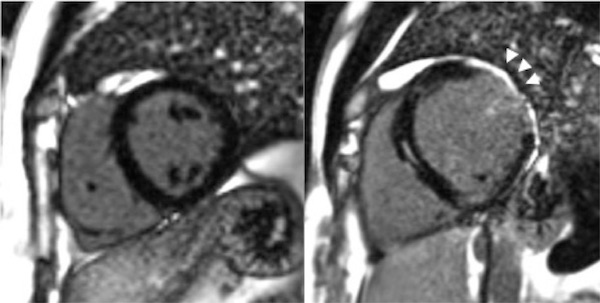
High levels of PAI-1 promote fibrosis in multiple organs.30,31 Paradoxically complete deficiency of PAI-1 is associated with cardiac fibrosis in the mouse model.32,33 PAI-1 controls the cardiomyocyte TGF-β axis and its deficiency leads to fibrosis as demonstrated in the PAI-1 deficient murine cardiac injury model. This was also demonstrated in humans with homozygous state for PAI-1 deficiency.34 Seven of the ten homozygotes in the Amish kindred described above, aged 15-35 years were found to have cardiac fibrosis of varying degrees (1-19% on MRI); the individual with 19% fibrosis had a poor ejection fraction of 32% and had a sudden cardiac death (Figure 2).35 Evidence of cardiac abnormalities in evaluated homozygous PAI-1 patients emphasizes the need for screening echocardiograms annually (starting at age 15 years to assess ventricular function) and performing cardiac MRIs as needed to quantify percent fibrosis and progression. These are the first cases of fibrotic cardiomyopathy described in PAI-1 deficient patients. There is currently no specific treatment for cardiac fibrosis associated with complete PAI-1 deficiency; treatment is symptomatic.35,36
Figure 2. Cardiac fibrosis in a homozygous PAI-1 deficient patient (arrows). Images from Gupta S, Khan S, Shah S, et al. ABSTRACT PB 1996: Association of homozygous PAI-1 deficiency with cardiac fibrosis in humans. Research and Practice in Thrombosis and Haemostasis 2017;1:846.
Elevated PAI- 1: Elevated PAI-1 levels are observed in individuals with a specific polymorphism and are suggested to be related to development of venous thrombosis (VT). This polymorphism is a single guanine deletion/insertion polymorphism (4G/5G) in the promoter of the plasminogen activator inhibitor type 1 (PAI-1) gene, situated 675 base pairs upstream from the transcriptional start site (PAI-14G/5G). The 4G allele is associated with enhanced gene expression, resulting in increased plasma levels of PAI-1.37,38 Although a clear causal relationship between the 4G/5G polymorphism and the risk of VT is not clear,39-41 it is presumed that a sustained increase in PAI-1 plays a role in the pathogenesis of VT due to impaired fibrinolysis.
Elevated PAI-1 has also been linked with myocardial infarction,42,43 post-operative thrombosis following hip replacement,44 arterial thrombosis,45 and poor cancer prognosis.46 Glueck et al. (2001) reported an increased risk of severe pre-eclampsia, placental abruption, IUGR and still birth associated with high PAI-1 levels and the 4G/4G mutation of the PAI-1 gene.47 Increased levels of PAI-1 expression in breast cancer are associated with a poor prognosis.48
Incidence of PAI-1 deficiency: The incidence of PAI-1 deficiency is unknown. This is in part due to the lack of sensitive diagnostic assays and the rarity of the disease. A survey within the Federally Recognized Hemophilia Treatment Centers in the United States revealed few patients with documented PAI-1 deficiency.49 Another study compared 586 individuals known to have a bleeding tendency with 200 controls and found that 23% of the individuals with a bleeding tendency had low PAI-1 activity as compared with 10-13% of controls.50 When 66 blood donors were evaluated, 14.6% of the females and 21% of the males had a PAI-1 activity < 2 IU/ml.51 As the number of case reports of PAI-1 deficiency are small, true incidence and prevalence cannot be determined.
Influence of race, sex and age: Beyond certain groups where consanguineous unions are more frequent, there is no known racial or ethnic predilection for this rare bleeding disorder. Males and females are equally affected; women may either present with clinical symptoms more frequently or earlier in life due menorrhagia and post-partum hemorrhage. Age of presentation is extremely variable. As these inferences are made from a small sample size of cases in the literature, their accuracy is difficult to assess. Cases of PAI-1 deficiency have been reported in North America, Europe and Asia.
History of PAI-1 deficiency: The first published link between an altered PAI-activity associated with a hemorrhagic tendency was reported in 1989.52 The report documented a lifelong history of severe bleeding following surgery or trauma and evidence of hyperfibrinolysis in an elderly man. Upon analysis, it was demonstrated that his PAI-1 antigen levels were normal with reduced PAI-1 activity, likely representing a qualitative defect. Additional cases of qualitative PAI-1 deficiency have subsequently been identified.20,53-59
The first quantitative PAI-1 defect was described in 1991 and detailed a 36 year old female with lifelong epistaxis and a history of bleeding after minor surgery.60 She had no detectable PAI-1 antigen or activity. The first genetic defect linking a bleeding diathesis to a homozygous PAI-1 mutation was reported in 1992.53 Since that time, two other specific genetic mutations have been identified that are associated with the bleeding disorder.59,61,62 (Please refer to the ‘Genetic defects’ section for more detailed information).
Genetic defects: At this time, there are four genetic defects known to be associated with PAI-1 deficiency. The first genetic defect was discovered in 1992 in a nine year old girl from an Old Order Amish community.53 She had a history of major hemorrhage in response to surgery or trauma and was found to be homozygous for a dinucleotide insertion in exon 4 of the PAI-1 gene. The insertion resulted in a frame-shift mutation which led to the formation of a premature stop codon and synthesis of a truncated, non-functional protein. The mutation is identified as SERPINE1 (c.699_700dupTA). Subsequently, a report from a more extensive kindred analysis revealed six additional members with the same homozygous mutation; all displaying mild-moderate bleeding symptoms.20 Ninety six individuals within this community harboring a heterozygous mutation did not display bleeding symptoms (unpublished data, Indiana Hemophilia and Thrombosis Center, [IHTC]). After a recent NIH funded study conducted in this population in 2015, additional patients have been identified resulting in a total of 108 heterozygotes and 11 homozygotes. A PAI-1 deficient 32 year old male is deceased due to sudden cardiac death.35 Although PAI-1 plays an important role in several biological processes other than fibrinolysis as mentioned above, it is surprising that these reported cases did not display physical or developmental abnormalities.
The second genetic mutation leading to PAI-1 deficiency was found in a 34-year old Chinese male with a life-long history of bleeding associated with surgery or trauma. A heterozygous missense mutation in exon 2 was discovered which led to the replacement of alanine to threonine at codon 15 of the signal peptide. Of interest is the observation that the patient’s father was heterozygous for the same mutation but did not exhibit bleeding symptoms, while his mother had moderately reduced PAI-1 levels. The authors of the paper concluded that this patient displayed bleeding symptoms likely due to a compound heterozygous state with the known mutation in exon 2 being only partially responsible for low PAI-1 levels.59
A third mutation responsible for PAI-1 deficiency was found in a 47-year old female with a history of major bleeding through her entire life including omphalorrhagia at birth, prolonged bleeding after surgery, extreme menorrhagia with the loss of 6 L of blood during her first menstruation, antenatal and post-partum bleeding and miscarriage. She also had impaired wound healing. Upon genetic analysis, she was found to be homozygous for one base pair duplication on exon 3. Specifically, the duplication caused a frameshift mutation leading to formation of a premature stop codon and thus a truncated, nonfunctional protein.61,62
Most recently, Iwaki et al. (2017) described a 70-year female with functional deficiency of PAI-1. She had a life time bleeding history and was found to have a homozygous single nucleotide substitution from guanine to cytosine at exon 9 which changes amino acid residue 397 from glycine to arginine, c.1189G>C;p.Gly397Arg. Her bleeding symptoms included uterine hemorrhage with pregnancy, subdural cerebellar hemorrhage, menorrhagia and bleeding after tooth extraction.63
Acquired deficiency of PAI-1: To-date, there is one reported case of acquired PAI-1 deficiency. The report details a 49-year old male with end-stage liver disease who suffered spontaneous, life-threatening, deep muscle bleeding on several occasions. Upon laboratory analysis, it was determined that he had undetectable PAI-1 activity, detectable PAI-1 antigen and high t-PA antigen levels and activity. His fibrinolytic activity was attributed to lack of clearance of t-PA by the dysfunctional liver with acquired relative PAI-1 deficiency. Antifibrinolytic therapy prevented significant bleeding episodes and PAI-1 activity normalized three months post liver transplant.64 In another case, elevated t-PA levels in the urine after trans-uretheral prostatectomy have been documented to occur along with increased blood loss post-procedure. Although PAI-1 activity was not determined in this case, it was proposed that elevated t-PA led to a locally acquired PAI-1 deficiency.65 Neutralizing antibodies to PAI-1 have been detected in one patient with amyloidosis, accelerated fibrinolysis and bleeding symptoms, which could be an explanation for decreased PAI-1 activity with normal t-PA antigen.66
