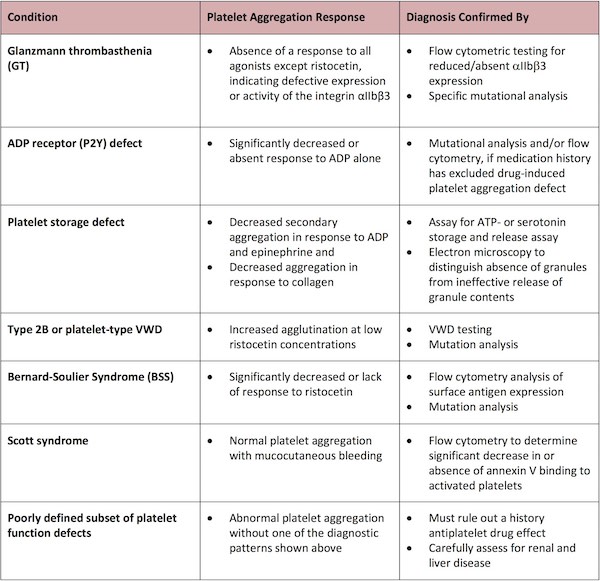
Certain patterns of platelet aggregation responses can establish a diagnosis. The finding of a kindred with a comparable aggregation defect can establish heritability of the disorder. Morphologic changes in the blood smear suggestive of myelodysplastic syndrome should be considered and if indicated, a bone marrow (BM) examination may be required to establish a diagnosis.
In the interim, it is hoped that ongoing genome-wide and exon sequencing studies of such inherited platelet disorders will permit the identification of novel genetic variants that contribute to the regulation of platelet function.
Absence of a response to all agonists except ristocetin is characteristic of a defect in expression or activity of the integrin aIIbb3 and can be confirmed by flow cytometric testing for reduced/absent aIIbb3 expression and specific mutational analysis to identify the genetic defect as Glanzmann thrombasthenia (GT).
A significantly decreased or absent response to ADP alone can indicate an ADP receptor (P2Y) defect, whereupon mutational analysis and/or flow cytometry can be employed to confirm this diagnosis, provided that a careful medication history has excluded an alternative diagnosis of a drug-induced platelet aggregation defect.
The finding of decreased secondary aggregation in response to ADP and epinephrine as well as decreased aggregation in response to collagen is most times indicative of a platelet storage defect, which can then be confirmed in most cases by an assay for ATP- or serotonin storage and release assay. Electron microscopy can also be used to distinguish the absence of granules from ineffective release of granule contents.
A finding of increased agglutination at low concentrations of ristocetin suggests the diagnoses of type 2B or platelet-type von Willebrand disease (VWD), which can be readily confirmed by VWD testing and mutation analysis.
The significantly decreased or absent response to ristocetin is typical of Bernard-Soulier Syndrome (BSS), which can then be confirmed by flow cytometry analysis of surface antigen expression and mutation analysis.
The finding of normal platelet aggregation in the face of mucocutaneous bleeding is also suggestive of Scott syndrome, a defect in platelet prothrombinase activity caused by a defective scramblase, which results in the inability to flip phosphatidylserine to the platelet surface upon activation.6 Scott syndrome can be diagnosed by the significant decrease in, or absence of, Annexin V binding to activated platelets, determined for example by flow cytometry.
The finding of abnormal platelet aggregation without one of these diagnostic patterns typifies a subset of poorly defined platelet function defects. In such cases, it is imperative to rule out a history antiplatelet drug effects and to perform a careful assessment for renal and liver disease.
Table 2 summarizes the platelet aggregation responses that indicate specific platelet defects and the methods used to confirm the diagnosis.