
VKCFD1 results from autosomal recessive inheritance of mutation in the GGCX gene. Thirty-three individual GGCX mutations (21 distinct genotypes) have been identified in the 33 reported VKCFD1 subjects (see previous paragraph); 14/33 carried homozygous mutations. Compound heterozygous defects have been documented in the remaining affected individuals. 29,30,31 The very recent genetic characterization of two unique GGCX mutations in the original VKCFD index case described by McMillan and Roberts increases the number to 35 mutations.7 Almost half of the characterized mutations fall in the region of AA 56-316 that includes the first four transmembrane domains. A quarter of the mutations lie within or near the AA 491-507 propeptide binding site and 3 mutations involve the AA 393-404 binding site for the Glu of the substrate VKDPs. 5
Evaluating the VKCFD patient in a more global way than simply managing the liver-derived clotting factors is essential to understanding best health care practice of the individual and to elucidating the pathogenesis of undercarboxylation in both hepatic and extrahepatic tissues. Dasi et al., best illustrates this by the characterization of one exceptional case: VKCFD1 presenting as intramuscular bleeding post-vaccination in an infant subsequently demonstrated a homozygous mutation in the last nucleotide of the GGCX intron 1 (c.44-1G>A).23 The mutation is predicted to severely disrupt the acceptor sequence of intron 1. Parental examination revealed that the mother carried no GGCX mutation, and single nucleotide polymorphism (SNP) analysis demonstrated loss of heterozygosity (LOH) in the proband and uniparental disomy, with 2 copies of the paternal chromosome 2 (including the GGCX locus). The proband was further shown to express no complete transcript or protein product from the full-length GGCX gene, as expected, but did express a small isoform 2 mRNA transcript without an exon 2 (Δ2GGCX). Interestingly, vitamin K1 supplementation resulted in minimal change in the carboxylation and activity of VKDP clotting factors but very substantially corrected the carboxylation of the non-hepatic VKDP osteocalcin, potentially through enzymatic activity retained by the Δ2GGCX protein product. These results suggest that different domains of the GGCX protein (and, therefore, also mutations in different domains of the GGCX protein) may be associated with differential carboxylation of the various hepatic and non-hepatic VKDP, as well as the effect of vitamin K supplementation. An additional case recently described by Tie, et al., supports this same paradigm. In the latter example, a homozygous GGCX p.D153G mutation is shown to result in decreased activity of hepatic and non-hepatic VKDP, however, vitamin K supplementation in the case of this mutation resulted in up to 60% correction of coagulation factor carboxylation but did not change MGP undercarboxylation.22 These examples are offered to demonstrate that careful evaluation of genotype-phenotype correlations, ideally matched with testing of hepatic and non-hepatic VKDP and response to supplementation with vitamin K1, are dually informative in advancing mechanistic understanding of the GGCX protein and individual patient management. Further evolving insights regarding genotype-phenotype correlations are discussed in the section “Clinical Manifestations”.24
In contrast to VKCFD1, one causative mutation in the VKOR gene underlies VKCFD2. It is striking that a single VKOR mutation has been associated with VKCFD2, because dozens of VKORC1 missense mutations are described and are associated not with VKCFD2, but instead with oral anticoagulant (warfarin) resistance.32 Homozygous mutation of VKORC1 nucleotide 292C→T results in substitution of tryptophan for arginine at amino acid 98.8,13 Also in contrast to VKCFD1, the pathogenesis resulting from the VKOR 292C→T mutation does not result from defective VKORC1 synthesis or abnormal interaction with the VKDP substrate. Instead, while the 100% of wild type VKORC1 is normally localized to the site of enzymatic action in the ER, the causative mutation at AA 98 disrupts the putative di-arginine ER retention motif.8,25 Expression of the variant Arg98Trp VKOR in in vitro systems suggests that while the majority of the VKOR is lost in the cytoplasm, a minor population remains localized in the ER. It is possible that the remaining ER Arg98Trp VKORC1 variant accounts for the correction of the VKCFD2 phenotype that has been generally achieved under daily vitamin K1 supplementation in vivo (see “Medications/Treatment” section).
Genetic testing is offered by the academic laboratories of Drs. Johannes Oldenburg, Jian-Ke Tie, and Flora Peyvandi.
Prof. Dr. Johannes Oldenburg Institute of Experimental Haematology and Transfusion Medicine University Clinic Bonn Sigmund-Freud-Str. 25 53127 Bonn, Germany Email: www.ukb.uni-bonn.de/iht
Jian-Ke Tie, PhD University of North Carolina at Chapel Hill Chapel Hill, NC Email: jktie@email.unc.edu Website: http://bio.unc.edu/people/faculty/tie-jianke/
Flora Peyvandi, MD Angelo Bianchi Bonomi Hemophilia and Thrombosis Center Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico And Department of Pathophysiology and Transplantation Università degli Studi di Milano 20122 – Milan, Italy Tel: +39 02 5503 5414 Fax: +39 02 54 100 125 Email: flora.peyvandi@unimi.it
For commercial laboratories that offer fee-for-service genetic testing, visit the Genetic Testing Registry website at https://www.ncbi.nlm.nih.gov/gtr/ (search “VKCFD”). There are currently 9 sites that offer testing.
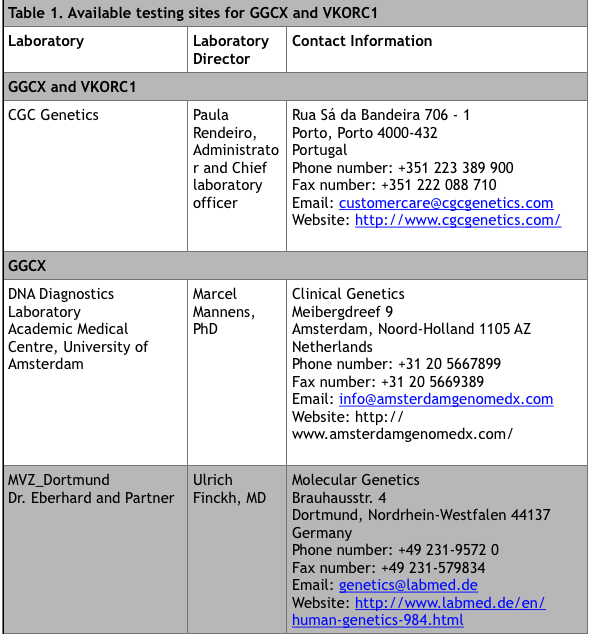
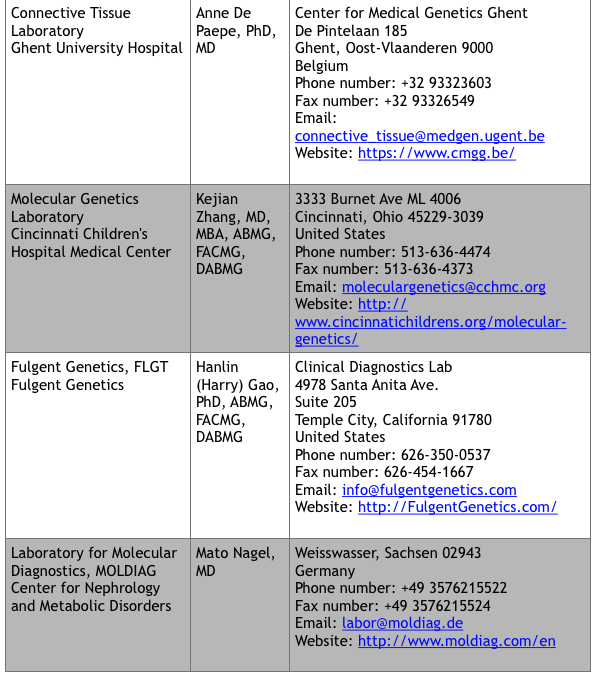
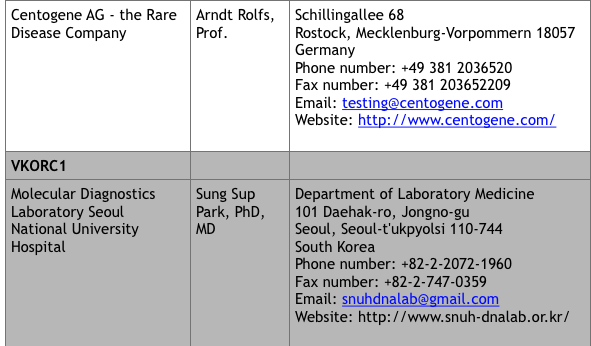
*Up to date at time of publication
