
Combined deficiency of vitamin K-dependent clotting factors (VKCFD) arises only rarely as a congenital condition. Congenital VKCFD is inherited as one of two variants; both are rare autosomal recessive bleeding disorders. The more common clinical presentation of combined deficient activity of clotting factors II (FII), FVII, FIX and FX (and proteins C, S and Z) occurs due to acquired vitamin K deficiency, in particular resulting from liver disease, malabsorption or malnutrition, or associated with a number of medications, most notably overdose of warfarin or warfarin derivatives.
The function of each of the vitamin K-dependent proteins (VKDP) depends upon normal gamma (Ƴ)-carboxylation of a number of glutamic acid residues in the amino-terminal region of the proteins (the Gla Domain).1 This reaction requires normal cycling of vitamin K from its reduced form to the oxidized state, followed by regeneration of reduced vitamin K.2, 3 These reactions are accomplished via the action of two endoplasmic reticulum (ER) enzymes: the Ƴ-glutamyl carboxylase (GGCX) and the vitamin K 2,3 epoxide reductase complex subunit1 (VKORC1). The two variants of VKCFD arise due to defects in the genes that encode these two proteins and not due to intrinsic defects in the clotting factors. Deficient activity of GGCX results in VKCFD1 while deficient activity of VKORC1 leads to VKCFD2. The most common clinical presentation of VKCFD is severe hemorrhage during infancy, although the degree of deficiency is highly variable and presentation of VKCFD may occur throughout childhood and in adulthood.4,5 Moreover, there is growing recognition that undercarboxylation and deficient function of VKDP affecting biological systems outside of coagulation is common in VKCFD, is highly variable in presentation, and may present clinical features considerably later in life than the bleeding phenotype, with implications for surveillance and management of VKCFD.
McMillan and Roberts described the first case of clinical hemorrhage resulting from congenitally deficient Ƴ-carboxylation in 1966.6,7 The female infant of an uncomplicated pregnancy manifested bruising beginning at age 1 week, with recurrent serosanguinous oozing from the umbilical stump throughout the first months of life. At presentation her prothrombin time (PT) was 95 seconds and her partial thromboplastin time (PTT) was 305 seconds. Upon mixing the patient’s plasma in a 1:1 ratio with normal plasma, each of these tests corrected completely to the normal range and clinically her umbilical bleeding stopped after a single infusion of fresh frozen plasma (FFP). While her platelet count, fibrinogen, and FV and FVIII were normal, activity of FII, FVII, FIX and FX were all below the lower limit of detection (<3%). On institution of a program of daily enteral vitamin K1 therapy, the child grew and developed normally with only a mild bruising tendency. An interruption of vitamin K1 therapy for a 9-day period resulted in the return of marked severe bruising.
During the ensuing half-century, diagnoses of VKCFD have been extremely rare. The most comprehensive collection of reported cases of VKCFD1 (genetic defect of GGCX associated with low VKDP clotting factors) includes fewer than 30 kindreds; 5,7 four families with VKCFD2 have been described.8 The degree of deficiency is variable among the reported cases. The particular combination of deficient FII, FVII, FIX and FX results in multiple levels of defective thrombin generation. Activity of the substrate prothrombin is itself deficient, and additionally thrombin generation via both the Tissue Factor/FVIIa and via the tenase complex is impaired. Deficiency of proteins in each of the intrinsic, extrinsic, and common pathways of the coagulation cascade is reflected in prolongation of both the PT and aPTT screening tests. The prolongation of the PT may be more marked than the prolongation of the aPTT in milder cases. Ƴ-carboxylation is required also for the normal function of the anticoagulant proteins C, S, and Z; however, the common presentation and severe complications of VKCFD result from bleeding rather than thrombosis.9
The full etiology of VKCFD became clear after the cloning and characterization of the gene encoding the GGCX protein by Stafford and colleagues in 199110,11 Stafford and colleagues and Oldenburg and colleagues in 2004.12,13,14 Subsequent pedigree studies have led to the designation of the two variants: VKCFD1, resulting from mutations in the Ƴ-glutamyl carboxylase gene (GGCX), and VKCFD2, resulting from mutations in the VKOR complex gene (VKORC1, or VKOR).15,16,17
Vitamin K exists in 3 forms:
The vitamin K dependent proteins all contain a Ƴ-carboxyglutamic acid-rich Gla domain, and the role of vitamin K as a co-factor in the proper post-translational modification of this domain is critical to their normal function. In addition to the proteins involved in coagulation, it is increasingly appreciated that a variety of biological processes are affected by vitamin K dependent proteins (VKDP) and the phenotypic expression of under-carboxylation may be associated with bleeding and non-bleeding manifestations. (See “Clinical Manifestations.”) In addition to the involvement of the anti-coagulant proteins C and S in inflammation, the action of Gla-rich protein (GRP) in monocyte/macrophages is involved in the interplay between inflammation and calcification. The osteoblast-derived VKDP osteocalcin is involved in glucose metabolism and associated with normal bone formation and bone strength. Additional VKDP are involved in the regulation of cell proliferation (Growth Arrest Specific gene 6 protein, Gas6) and with the inhibition of vascular calcification and tissue mineralization (matrix Gla protein, MGP), while the function of several additional VKDP are currently unknown, including transmembrane Gla proteins (TMG) and proline-rich Gla proteins (PRGP).2,18, 19, 20
Clotting factors II, VII, IX and X and proteins C, S and Z contain anywhere from 9 to 13 glutamates (Glu) in the amino acid sequence of their respective Gla domains. Insufficient posttranslational modification of these glutamates to Ƴ-carboxyglutamic acid (Gla) is the enzymatic step that is defective in both variants of VKCFD. The carboxylation of the Gla domain confers a calcium-dependent conformation that is critical to normal interaction of the proteins with phospholipids (e.g., with the phospholipid surface of the activated platelet) or with endothelial cells. The GGCX is the protein that performs this posttranslational modification; however, vitamin K in its reduced form is required as well to act as a cofactor. Vitamin K reduced is converted to vitamin K epoxide in the course of the reaction, and the reduced form must be regenerated for additional cycles of catalysis via the action of VKOR. VKOR is opposed by the anticoagulant warfarin, so that an individual taking warfarin has impaired regeneration of vitamin K reduced, with a controlled result similar to vitamin K deficiency or VKCFD: excess undercarboxylated and functionally deficient FII, FVII, FIX, and FX. 2,3 The vitamin K cycles can be visualized as shown in Figure 1.
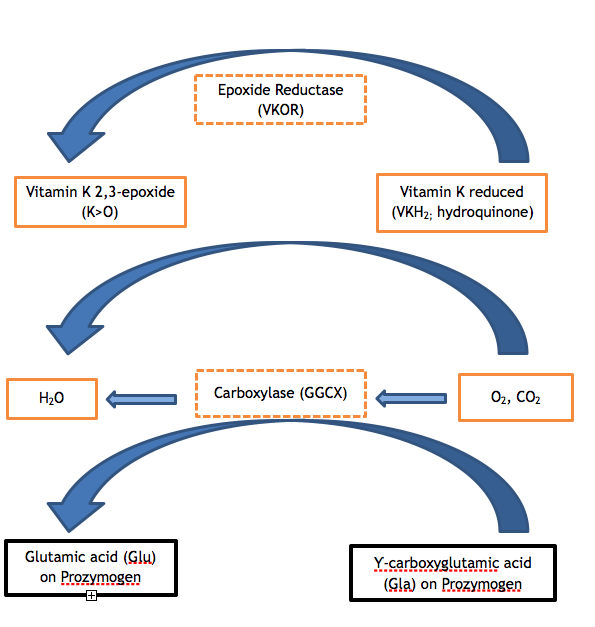
Figure 1. The Vitamin K Cycle. Carboxylation of glutamic acid residues of the zymogen vitamin K-dependent proteins is necessary for their proper function. The carboxylase (GGCX) adds a carbon dioxide molecule to Glu to form Gla in a reaction for which vitamin K is an essential co-factor. The reduced form of vitamin K (hydroquinone) is oxidized in the process, yielding vitamin K 2,3-epoxide. Vitamin K reduced must be regenerated from the epoxide by the enzymatic action of VKOR so that Vitamin K reduced is available to serve as a cofactor for subsequent carboxylation reactions. Deficient activity of either of the enzymes VKOR or GGCX (see hatched boxes in figure) results in VKCFD.
It is evident from this discussion that while VKDPs are synthesized and secreted normally, protein function is abnormal as a result of insufficient post-translational carboxylation.
The purification of GGCX and VKORC1, which are integral membrane proteins, has been challenging and the crystal structures of these proteins are not yet resolved. Nevertheless, an understanding of the functional domains of these proteins is emerging.1 This effort is complemented by recent efforts to correlate gene defects affecting specific regions of the enzymes with the severity and spectrum of deficient VKDP activity, both in the coagulation system and in other physiological processes. 5,8,21,22
The GGCX gene on the p arm of chromosome 2 codes for a 758 amino acid (AA) transmembrane protein that is localized on the membrane of the endoplasmic reticulum. The N-terminal portion of the enzyme is cytoplasmic. Five transmembrane domains follow, localized in the lipid membrane of the ER. The C-terminal portion of the protein is intralumenal in the ER. The intralumenal region contains hydrophobic domains important for interacting with vitamin K. An important functional domain (proposed to be AA 491-507) interacts with the propeptide of the VKDP, while another binding region (AA 393-404) for the VKDP glutamate facilitates gamma-carboxylation. In fact, the first correlation of the clinical phenotype of VKCFD with a mutation in the GGCX gene described a substitution of leucine for arginine at position 394 of the glutamate binding region.15 Although the sequence of the first four GGCX transmembrane domains is highly evolutionarily conserved, their function in humans is not known. The protein described here is expressed from the full length GGCX gene. An isoform 2 of the GGCX mRNA that lacks exon 2 (Δ2GGCX) may encode a protein that retains enzymatic activity, although the physiologic significance of this isoform is not determined (see also “Inheritance and Causative Mutations”).23
The chromosome 16 VKOR gene encodes a 163 AA transmembrane protein. The basic membrane topology of the VKOR protein is controversial: for example, debate continues whether the protein has three or four transmembrane domains, and whether the highly conserved VKOR C132XXC135 active site for the nucleophilic attack on Vitamin K is cytoplasmic or inside the ER lumen. An important structural region involves the arginine moieties at AA 98 and 100 constitute a di-arginine ER retention motif that is necessary to keep VKOR localized in the required site of activity. A single VKOR mutation (VKORC1p.Arg98Trp) accounts for all cases of VKCFD2, and so the opportunity does not exist for genotype/phenotype correlations to inform understanding of the functional enzymatic interactions of the reductase (analogous to the possibilities that exist with VKCFD1). 1,8,24,25
The possibility exists that additional genes that modify the action of these proteins may as yet be unidentified. Animal data suggests that an independent, redundant carboxylase pathway does not exist in all tissues, however, in light of the embryonic lethality of both GGCX knockout and VKOR knockout in mice.26,27 At the time of the identification of the VKOR protein, Rost and colleagues identified a paralogous protein labeled VKOR-like 1 (VKORL1), which is capable of reducing K>O;13 a role for VKORL1 to rescue some vitamin K function in extra-hepatic tissues during anticoagulation with vitamin K antagonists has been suggested.1 There is no data to suggest that VKORL1 has a significant physiologic role in supporting hepatic carboxylation of clotting factors, so the paralogous protein is not thought to be relevant to VKCFD at this time.
VKCFD is very rare. The most comprehensive collection of published cases of VKCFD1 (in which a gene mutation in GGCX is present in association with decreased coagulation factor activity) includes 33 affected individuals (26 separate probands).5 No influence of race or ethnicity has been described, and cases have been reported from Africa, Asia, Europe, North America and South America.7,21,28 VKCFD2 is more rare, with 10 reported cases from four families of North American Caucasian, German, Italian, and Lebanese origin.8
